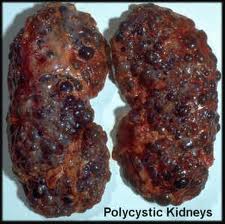
Relation to other rare genetic disorders
Recent findings in genetic research have suggested that a large number of genetic disorders, both genetic syndromes and genetic diseases, that were not previously identified in the medical literature as related, may be, in fact, highly related in the genetypical root cause of the widely-varying, phenotypically-observed disorders. Thus, PKD is a ciliopathy. Other known ciliopathies include primary ciliary dyskinesia, Bardet-Biedl syndrome, polycystic liver disease, nephronophthisis, Alstrom syndrome, Meckel-Gruber syndrome and some forms of retinal degeneration.
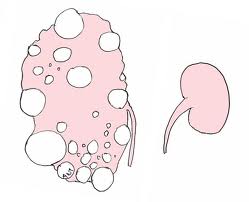
Right: Normal kidney. Left: Polycystic kidney
Symptoms
- Abdominal pain or tenderness
- Blood in the urine
- Excessive urination at night
- Flank pain on one or both sides
Other symptoms that may occur with this disease include:
- Drowsiness
- Joint pain
- Nail abnormalities
Signs and tests
An examination may show:
- Abdominal tenderness over the liver
- Enlarged liver
- Heart murmurs or other signs of aortic insufficiency or mitral insufficiency
- High blood pressure
- Growths in the kidneys or abdomen
Tests that may be done include:
- Cerebral angiography
- Complete blood count (CBC)
- Urinalysis
People with a personal or family history of PKD should be tested to determine if cerebral aneurysms are causing headaches.
Polycystic kidney disease and cysts on the liver or other organs may be found with the following tests:
- Abdominal CT scan
- Abdominal MRI scan
- Abdominal ultrasound
- Intravenous pyelogram (IVP)
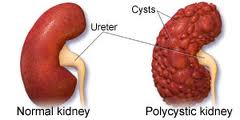 If several members of your family have PKD, genetic tests can be done to determine whether you carry the PKD gene.
If several members of your family have PKD, genetic tests can be done to determine whether you carry the PKD gene.
A definite diagnosis of ADPKD relies on imaging or molecular genetic testing. The sensitivity of testing is nearly 100% for all patients with ADPKD who are age 30 years or older and for younger patients with PKD1 mutations; these criteria are only 67% sensitive for patients with PKD2 mutations who are younger than age 30 years. Large echogenic kidneys without distinct macroscopic cysts in an infant/child at 50% risk for ADPKD are diagnostic. In the absence of a family history of ADPKD, the presence of bilateral renal enlargement and cysts, with or without the presence of hepatic cysts, and the absence of other manifestations suggestive of a different renal cystic disease provide presumptive, but not definite, evidence for the diagnosis.
Molecular genetic testing by linkage analysis or direct mutation screening is available clinically; however, genetic heterogeneity is a significant complication to molecular genetic testing. Sometimes a relatively large number of affected family members need to be tested in order to establish which one of the two possible genes is responsible within each family. The large size and complexity of PKD1 and PKD2 genes, as well as marked allelic heterogeneity, present obstacles to molecular testing by direct DNA analysis. In the research setting, mutation detection rates of 50-75% have been obtained for PKD1 and ~75% for PKD2. Clinical testing of the PKD1 and PKD2 genes by direct sequence analysis is now available, with a detection rate for disease-causing mutations of 50-70%.
Genetic counseling may be helpful for families at risk for polycystic kidney disease.
Treatment
The goal of treatment is to control symptoms and prevent complications. High blood pressure may be hard to control, but controlling it is the most important part of treatment.
Treatment may include:
- Blood pressure medicines
- Diuretics
- Low-salt diet
Any urinary tract infection should be treated quickly with antibiotics.
Cysts that are painful, infected, bleeding, or causing a blockage may need to be drained. There are usually too many cysts to make it practical to remove each cyst.
Surgery to remove one or both kidneys may be needed. Treatments for end-stage kidney disease may include dialysis or a kidney transplant.
Support Groups
You can often ease the stress of an illness by joining a support group where members share common experiences and problems.
Expectations (prognosis)
Despite significant research, prognosis of this disease has changed little over time. It is suggested that avoidance of caffeine may prevent cyst formation. Although not well-proven, treatment of hypertension and a low protein diet may slow progression of the disease.
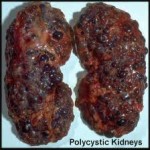 Between PKD1 and PKD2, the former has the worse prognosis.
Between PKD1 and PKD2, the former has the worse prognosis.
The disease gets worse slowly. Eventually it leads to end-stage kidney failure. It is also associated with liver disease, including infection of liver cysts.
Medical treatment may relieve symptoms for many years.
People with PKD who don’t have other diseases may be good candidates for a kidney transplant.
Complications
- Anemia
- Bleeding or rupture of cysts
- Chronic kidney disease
- End-stage kidney disease
- High blood pressure
- Infection of liver cysts
- Kidney stones
- Liver failure (mild to severe)
- Repeated urinary tract infections
Calling your health care provider
Call your health care provider if:
- You have symptoms of PKD
- You have a family history of polycystic kidney disease or related disorders and you are planning to have children (you may want to have genetic counseling)
Prevention
Currently, no treatment can prevent the cysts from forming or enlarging.